BLAST (new)
Hey, everybody loves BLAST right? I mean, geez, how can it get any easier to do comparisons between one of your sequences and every other sequence in the known world? But, of course, this section isn’t about how cool BLAST is, since we already know that. It is about the problem with BLAST – it can be really difficult to deal with the volume of data generated by large runs, and to automate BLAST runs in general.
Fortunately, the Biopython folks know this only too well, so they’ve developed lots of tools for dealing with BLAST and making things much easier. This section details how to use these tools and do useful things with them.
Dealing with BLAST can be split up into two steps, both of which can be done from within Biopython. Firstly, running BLAST for your query sequence(s), and getting some output. Secondly, parsing the BLAST output in Python for further analysis.
Your first introduction to running BLAST was probably via the NCBI BLAST web page. In fact, there are lots of ways you can run BLAST, which can be categorized in several ways. The most important distinction is running BLAST locally (on your own machine), and running BLAST remotely (on another machine, typically the NCBI servers). We’re going to start this chapter by invoking the NCBI online BLAST service from within a Python script.
Running BLAST over the Internet
We use the function qblast in the Bio.Blast module to call the
online version of BLAST.
The NCBI guidelines state:
Do not contact the server more often than once every 10 seconds.
Do not poll for any single RID more often than once a minute.
Use the URL parameter email and tool, so that the NCBI can contact you if there is a problem.
Run scripts weekends or between 9 pm and 5 am Eastern time on weekdays if more than 50 searches will be submitted.
Blast.qblast follows the first two points automatically. To fulfill
the third point, set the Blast.email variable (the Blast.tool
variable is already set to "biopython" by default):
>>> from Bio import Blast
>>> Blast.tool
'biopython'
>>> Blast.email = "A.N.Other@example.com"
BLAST arguments
The qblast function has three non-optional arguments:
The first argument is the BLAST program to use for the search, as a lower case string. The programs and their options are described at the NCBI BLAST web page. Currently
qblastonly works with blastn, blastp, blastx, tblast and tblastx.The second argument specifies the databases to search against. Again, the options for this are available on NCBI’s BLAST Help pages.
The third argument is a string containing your query sequence. This can either be the sequence itself, the sequence in fasta format, or an identifier like a GI number.
The qblast function also takes a number of other option arguments,
which are basically analogous to the different parameters you can set on
the BLAST web page. We’ll just highlight a few of them here:
The argument
url_basesets the base URL for running BLAST over the internet. By default it connects to the NCBI.The
qblastfunction can return the BLAST results in various formats, which you can choose with the optionalformat_typekeyword:"XML","HTML","Text","XML2","JSON2", or"Tabular". The default is"XML", as that is the format expected by the parser, described in section Parsing BLAST output below.The argument
expectsets the expectation or e-value threshold.
For more about the optional BLAST arguments, we refer you to the NCBI’s own documentation, or that built into Biopython:
>>> from Bio import Blast
>>> help(Blast.qblast)
Note that the default settings on the NCBI BLAST website are not quite the same as the defaults on QBLAST. If you get different results, you’ll need to check the parameters (e.g., the expectation value threshold and the gap values).
For example, if you have a nucleotide sequence you want to search against the nucleotide database (nt) using BLASTN, and you know the GI number of your query sequence, you can use:
>>> from Bio import Blast
>>> result_stream = Blast.qblast("blastn", "nt", "8332116")
Alternatively, if we have our query sequence already in a FASTA formatted file, we just need to open the file and read in this record as a string, and use that as the query argument:
>>> from Bio import Blast
>>> fasta_string = open("m_cold.fasta").read()
>>> result_stream = Blast.qblast("blastn", "nt", fasta_string)
We could also have read in the FASTA file as a SeqRecord and then
supplied just the sequence itself:
>>> from Bio import Blast
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", "fasta")
>>> result_stream = Blast.qblast("blastn", "nt", record.seq)
Supplying just the sequence means that BLAST will assign an identifier
for your sequence automatically. You might prefer to call format on
the SeqRecord object to make a FASTA string (which will include the
existing identifier):
>>> from Bio import Blast
>>> from Bio import SeqIO
>>> records = SeqIO.parse("ls_orchid.gbk", "genbank")
>>> record = next(records)
>>> result_stream = Blast.qblast("blastn", "nt", format(record, "fasta"))
This approach makes more sense if you have your sequence(s) in a
non-FASTA file format which you can extract using Bio.SeqIO (see
Chapter Sequence Input/Output).
Saving BLAST results
Whatever arguments you give the qblast() function, you should get
back your results as a stream of bytes data (by default in XML
format). The next step would be to parse the XML output into Python
objects representing the search results
(Section Parsing BLAST output), but you might want to save a
local copy of the output file first. I find this especially useful when
debugging my code that extracts info from the BLAST results (because
re-running the online search is slow and wastes the NCBI computer time).
We need to be a bit careful since we can use result_stream.read() to
read the BLAST output only once – calling result_stream.read() again
returns an empty bytes object.
>>> with open("my_blast.xml", "wb") as out_stream:
... out_stream.write(result_stream.read())
...
>>> result_stream.close()
After doing this, the results are in the file my_blast.xml and
result_stream has had all its data extracted (so we closed it).
However, the parse function of the BLAST parser (described
in Parsing BLAST output) takes a file-like object, so we can
just open the saved file for input as bytes:
>>> result_stream = open("my_blast.xml", "rb")
Now that we’ve got the BLAST results back into a data stream again, we are ready to do something with them, so this leads us right into the parsing section (see Section Parsing BLAST output below). You may want to jump ahead to that now ….
Obtaining BLAST output in other formats
By using the format_type argument when calling qblast, you can
obtain BLAST output in formats other than XML. Below is an example of
reading BLAST output in JSON format. Using format_type="JSON2", the
data provided by Blast.qblast will be in zipped JSON format:
>>> from Bio import Blast
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", "fasta")
>>> result_stream = Blast.qblast("blastn", "nt", record.seq, format_type="JSON2")
>>> data = result_stream.read()
>>> data[:4]
b'PK\x03\x04'
which is the ZIP file magic number.
>>> with open("myzipfile.zip", "wb") as out_stream:
... out_stream.write(data)
...
13813
Note that we read and write the data as bytes. Now open the ZIP file
we created:
>>> import zipfile
>>> myzipfile = zipfile.ZipFile("myzipfile.zip")
>>> myzipfile.namelist()
['N5KN7UMJ013.json', 'N5KN7UMJ013_1.json']
>>> stream = myzipfile.open("N5KN7UMJ013.json")
>>> data = stream.read()
These data are bytes, so we need to decode them to get a string
object:
>>> data = data.decode()
>>> print(data)
{
"BlastJSON": [
{"File": "N5KN7UMJ013_1.json" }
]
}
Now open the second file contained in the ZIP file to get the BLAST results in JSON format:
>>> stream = myzipfile.open("N5KN7UMJ013_1.json")
>>> data = stream.read()
>>> len(data)
145707
>>> data = data.decode()
>>> print(data)
{
"BlastOutput2": {
"report": {
"program": "blastn",
"version": "BLASTN 2.14.1+",
"reference": "Stephen F. Altschul, Thomas L. Madden, Alejandro A. ...
"search_target": {
"db": "nt"
},
"params": {
"expect": 10,
"sc_match": 2,
"sc_mismatch": -3,
"gap_open": 5,
"gap_extend": 2,
"filter": "L;m;"
},
"results": {
"search": {
"query_id": "Query_69183",
"query_len": 1111,
"query_masking": [
{
"from": 797,
"to": 1110
}
],
"hits": [
{
"num": 1,
"description": [
{
"id": "gi|1219041180|ref|XM_021875076.1|",
...
We can use the JSON parser in Python’s standard library to convert the JSON data into a regular Python dictionary:
>>> import json
>>> d = json.loads(data)
>>> print(d)
{'BlastOutput2': {'report': {'program': 'blastn', 'version': 'BLASTN 2.14.1+',
'reference': 'Stephen F. Altschul, Thomas L. Madden, Alejandro A. Schäffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search programs",
Nucleic Acids Res. 25:3389-3402.',
'search_target': {'db': 'nt'}, 'params': {'expect': 10, 'sc_match': 2,
'sc_mismatch': -3, 'gap_open': 5, 'gap_extend': 2, 'filter': 'L;m;'},
'results': {'search': {'query_id': 'Query_128889', 'query_len': 1111,
'query_masking': [{'from': 797, 'to': 1110}], 'hits': [{'num': 1,
'description': [{'id': 'gi|1219041180|ref|XM_021875076.1|', 'accession':
'XM_021875076', 'title':
'PREDICTED: Chenopodium quinoa cold-regulated 413 plasma membrane protein 2-like (LOC110697660), mRNA',
'taxid': 63459, 'sciname': 'Chenopodium quinoa'}], 'len': 1173, 'hsps':
[{'num': 1, 'bit_score': 435.898, 'score': 482, 'evalue': 9.02832e-117,
'identity': 473, 'query_from'
...
Running BLAST locally
Introduction
Running BLAST locally (as opposed to over the internet, see Section Running BLAST over the Internet) has at least major two advantages:
Local BLAST may be faster than BLAST over the internet;
Local BLAST allows you to make your own database to search for sequences against.
Dealing with proprietary or unpublished sequence data can be another reason to run BLAST locally. You may not be allowed to redistribute the sequences, so submitting them to the NCBI as a BLAST query would not be an option.
Unfortunately, there are some major drawbacks too – installing all the bits and getting it setup right takes some effort:
Local BLAST requires command line tools to be installed.
Local BLAST requires (large) BLAST databases to be setup (and potentially kept up to date).
Standalone NCBI BLAST+
The “new” NCBI BLAST+ suite was released in 2009. This replaces the old NCBI “legacy” BLAST package (see Other versions of BLAST).
This section will show briefly how to use these tools from within Python. If you have already read or tried the alignment tool examples in Section Alignment Tools this should all seem quite straightforward. First, we construct a command line string (as you would type in at the command line prompt if running standalone BLAST by hand). Then we can execute this command from within Python.
For example, taking a FASTA file of gene nucleotide sequences, you might want to run a BLASTX (translation) search against the non-redundant (NR) protein database. Assuming you (or your systems administrator) has downloaded and installed the NR database, you might run:
$ blastx -query opuntia.fasta -db nr -out opuntia.xml -evalue 0.001 -outfmt 5
This should run BLASTX against the NR database, using an expectation cut-off value of \(0.001\) and produce XML output to the specified file (which we can then parse). On my computer this takes about six minutes - a good reason to save the output to a file so you can repeat any analysis as needed.
From within python we can use the subprocess module to build the
command line string, and run it:
>>> import subprocess
>>> cmd = "blastx -query opuntia.fasta -db nr -out opuntia.xml"
>>> cmd += " -evalue 0.001 -outfmt 5"
>>> subprocess.run(cmd, shell=True)
In this example there shouldn’t be any output from BLASTX to the
terminal. You may want to check the output file opuntia.xml has been
created.
As you may recall from earlier examples in the tutorial, the
opuntia.fasta contains seven sequences, so the BLAST XML output
should contain multiple results. Therefore use Bio.Blast.parse() to
parse it as described below in Section Parsing BLAST output.
Other versions of BLAST
NCBI BLAST+ (written in C++) was first released in 2009 as a replacement
for the original NCBI “legacy” BLAST (written in C) which is no longer
being updated. You may also come across Washington University
BLAST (WU-BLAST), and its successor,
Advanced Biocomputing BLAST
(AB-BLAST, released in 2009, not free/open source). These packages
include the command line tools wu-blastall and ab-blastall,
which mimicked blastall from the NCBI “legacy” BLAST suite.
Biopython does not currently provide wrappers for calling these tools,
but should be able to parse any NCBI compatible output from them.
Parsing BLAST output
As mentioned above, BLAST can generate output in various formats, such as XML,
HTML, and plain text. Originally, Biopython had parsers for BLAST plain text
and HTML output, as these were the only output formats offered at the time.
These parsers have now been removed from Biopython, as the BLAST output in
these formats kept changing, each time breaking the Biopython parsers.
Nowadays, Biopython can parse BLAST output in the XML format, the XML2 format,
and tabular format. This chapter describes the parser for BLAST output in the
XML and XML2 formats using the Bio.Blast.parse function. This function
automatically detects if the XML file is in the XML format or in the XML2
format.
BLAST output in tabular format can be parsed as alignments using the
Bio.Align.parse function (see the section Tabular output from BLAST or FASTA).
You can get BLAST output in XML format in various ways. For the parser, it doesn’t matter how the output was generated, as long as it is in the XML format.
You can use Biopython to run BLAST over the internet, as described in section Running BLAST over the Internet.
You can use Biopython to run BLAST locally, as described in section Running BLAST locally.
You can do the BLAST search yourself on the NCBI site through your web browser, and then save the results. You need to choose XML as the format in which to receive the results, and save the final BLAST page you get (you know, the one with all of the interesting results!) to a file.
You can also run BLAST locally without using Biopython, and save the output in a file. Again, you need to choose XML as the format in which to receive the results.
The important point is that you do not have to use Biopython scripts to
fetch the data in order to be able to parse it. Doing things in one of
these ways, you then need to get a file-like object to the results. In
Python, a file-like object or handle is just a nice general way of
describing input to any info source so that the info can be retrieved
using read() and readline() functions (see
Section What the heck is a handle?).
If you followed the code above for interacting with BLAST through a
script, then you already have result_stream, the file-like object to
the BLAST results. For example, using a GI number to do an online
search:
>>> from Bio import Blast
>>> result_stream = Blast.qblast("blastn", "nt", "8332116")
If instead you ran BLAST some other way, and have the BLAST output (in
XML format) in the file my_blast.xml, all you need to do is to open
the file for reading (as bytes):
>>> result_stream = open("my_blast.xml", "rb")
Now that we’ve got a data stream, we are ready to parse the output. The code to parse it is really quite small. If you expect a single BLAST result (i.e., you used a single query):
>>> from Bio import Blast
>>> blast_record = Blast.read(result_stream)
or, if you have lots of results (i.e., multiple query sequences):
>>> from Bio import Blast
>>> blast_records = Blast.parse(result_stream)
Just like Bio.SeqIO and Bio.Align (see
Chapters Sequence Input/Output
and Sequence alignments), we have a pair of input
functions, read and parse, where read is for when you have
exactly one object, and parse is an iterator for when you can have
lots of objects – but instead of getting SeqRecord or Alignment
objects, we get BLAST record objects.
To be able to handle the situation where the BLAST file may be huge,
containing thousands of results, Blast.parse() returns an iterator.
In plain English, an iterator allows you to step through the BLAST
output, retrieving BLAST records one by one for each BLAST search
result:
>>> from Bio import Blast
>>> blast_records = Blast.parse(result_stream)
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
StopIteration
# No further records
Or, you can use a for-loop:
>>> for blast_record in blast_records:
... pass # Do something with blast_record
...
Note though that you can step through the BLAST records only once. Usually, from each BLAST record you would save the information that you are interested in.
Alternatively, you can use blast_records as a list, for example by
extracting one record by index, or by calling len or print on
blast_records. The parser will then automatically iterate over the records
and store them:
>>> from Bio import Blast
>>> blast_records = Blast.parse("xml_2222_blastx_001.xml")
>>> len(blast_records) # this causes the parser to iterate over all records
7
>>> blast_records[2].query.description
'gi|5690369|gb|AF158246.1|AF158246 Cricetulus griseus glucose phosphate isomerase (GPI) gene, partial intron sequence'
If your BLAST file is huge though, you may run into memory problems trying to save them all in a list.
If you start iterating over the records before using blast_records as
a list, the parser will first reset the file stream to the beginning of the
data to ensure that all records are neing read. Note that this will fail if the
stream cannot be reset to the beginning, for example if the data are being
read remotely (e.g. by qblast; see subsection Running BLAST over the Internet).
In those cases, you can explicitly read the records into a list by calling
blast_records = blast_records[:] before iterating over them. After reading
in the records, it is safe to iterate over them or use them as a list.
Instead of opening the file yourself, you can just provide the file name:
>>> from Bio import Blast
>>> with Blast.parse("my_blast.xml") as blast_records:
... for blast_record in blast_records:
... pass # Do something with blast_record
...
In this case, Biopython opens the file for you, and closes it as soon as
the file is not needed any more (while it is possible to simply use
blast_records = Blast.parse("my_blast.xml"), it has the disadvantage
that the file may stay open longer than strictly necessary, thereby
wasting resources).
You can print the records to get a quick overview of their contents:
>>> from Bio import Blast
>>> with Blast.parse("my_blast.xml") as blast_records:
... print(blast_records)
...
Program: BLASTN 2.2.27+
db: refseq_rna
Query: 42291 (length=61)
mystery_seq
Hits: ---- ----- ----------------------------------------------------------
# # HSP ID + description
---- ----- ----------------------------------------------------------
0 1 gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 52...
1 1 gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA...
2 1 gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA ...
3 2 gi|301171322|ref|NR_035857.1| Pan troglodytes microRNA...
4 1 gi|301171267|ref|NR_035851.1| Pan troglodytes microRNA...
5 2 gi|262205330|ref|NR_030198.1| Homo sapiens microRNA 52...
6 1 gi|262205302|ref|NR_030191.1| Homo sapiens microRNA 51...
7 1 gi|301171259|ref|NR_035850.1| Pan troglodytes microRNA...
8 1 gi|262205451|ref|NR_030222.1| Homo sapiens microRNA 51...
9 2 gi|301171447|ref|NR_035871.1| Pan troglodytes microRNA...
10 1 gi|301171276|ref|NR_035852.1| Pan troglodytes microRNA...
11 1 gi|262205290|ref|NR_030188.1| Homo sapiens microRNA 51...
12 1 gi|301171354|ref|NR_035860.1| Pan troglodytes microRNA...
13 1 gi|262205281|ref|NR_030186.1| Homo sapiens microRNA 52...
14 2 gi|262205298|ref|NR_030190.1| Homo sapiens microRNA 52...
15 1 gi|301171394|ref|NR_035865.1| Pan troglodytes microRNA...
16 1 gi|262205429|ref|NR_030218.1| Homo sapiens microRNA 51...
17 1 gi|262205423|ref|NR_030217.1| Homo sapiens microRNA 52...
18 1 gi|301171401|ref|NR_035866.1| Pan troglodytes microRNA...
19 1 gi|270133247|ref|NR_032574.1| Macaca mulatta microRNA ...
20 1 gi|262205309|ref|NR_030193.1| Homo sapiens microRNA 52...
21 2 gi|270132717|ref|NR_032716.1| Macaca mulatta microRNA ...
22 2 gi|301171437|ref|NR_035870.1| Pan troglodytes microRNA...
23 2 gi|270133306|ref|NR_032587.1| Macaca mulatta microRNA ...
24 2 gi|301171428|ref|NR_035869.1| Pan troglodytes microRNA...
25 1 gi|301171211|ref|NR_035845.1| Pan troglodytes microRNA...
26 2 gi|301171153|ref|NR_035838.1| Pan troglodytes microRNA...
27 2 gi|301171146|ref|NR_035837.1| Pan troglodytes microRNA...
28 2 gi|270133254|ref|NR_032575.1| Macaca mulatta microRNA ...
29 2 gi|262205445|ref|NR_030221.1| Homo sapiens microRNA 51...
~~~
97 1 gi|356517317|ref|XM_003527287.1| PREDICTED: Glycine ma...
98 1 gi|297814701|ref|XM_002875188.1| Arabidopsis lyrata su...
99 1 gi|397513516|ref|XM_003827011.1| PREDICTED: Pan panisc...
Usually, you’ll be running one BLAST search at a time. Then, all you
need to do is to pick up the first (and only) BLAST record in
blast_records:
>>> from Bio import Blast
>>> blast_records = Blast.parse("my_blast.xml")
>>> blast_record = next(blast_records)
or more elegantly:
>>> from Bio import Blast
>>> blast_record = Blast.read(result_stream)
or, equivalently,
>>> from Bio import Blast
>>> blast_record = Blast.read("my_blast.xml")
(here, you don’t need to use a with block as Blast.read will
read the whole file and close it immediately afterwards).
I guess by now you’re wondering what is in a BLAST record.
The BLAST Records, Record, and Hit classes
The BLAST Records class
A single BLAST output file can contain output from multiple BLAST queries. In
Biopython, the information in a BLAST output file is stored in an
Bio.Blast.Records object. This is an iterator returning one
Bio.Blast.Record object (see subsection The BLAST Record class) for
each query. The Bio.Blast.Records object has the following attributes describing the BLAST run:
source: The input data from which theBio.Blast.Recordsobject was constructed (this could be a file name or path, or a file-like object).program: The specific BLAST program that was used (e.g., ’blastn’).version: The version of the BLAST program (e.g., ’BLASTN 2.2.27+’).reference: The literature reference to the BLAST publication.db: The BLAST database against which the query was run (e.g., ’nr’).query: ASeqRecordobject which may contain some or all of the following information:query.id: SeqId of the query;query.description: Definition line of the query;query.seq: The query sequence.
param: A dictionary with the parameters used for the BLAST run. You may find the following keys in this dictionary:'matrix': the scoring matrix used in the BLAST run (e.g., ’BLOSUM62’) (string);'expect': threshold on the expected number of chance matches (float);'include': e-value threshold for inclusion in multipass model in psiblast (float);'sc-match': score for matching nucleotides (integer);'sc-mismatch': score for mismatched nucleotides (integer;'gap-open': gap opening cost (integer);'gap-extend': gap extension cost (integer);'filter': filtering options applied in the BLAST run (string);'pattern': PHI-BLAST pattern (string);'entrez-query': Limit of request to Entrez query (string).
mbstat: A dictionary with Mega BLAST search statistics. See the description of theRecord.statattribute below (in subsection The BLAST Record class) for a description of the items in this dictionary. Only older versions of Mega BLAST store this information. As it is stored near the end of the BLAST output file, this attribute can only be accessed after the file has been read completely (by iterating over the records until aStopIterationis issued).
For our example, we find:
>>> blast_records
<Bio.Blast.Records source='my_blast.xml' program='blastn' version='BLASTN 2.2.27+' db='refseq_rna'>
>>> blast_records.source
'my_blast.xml'
>>> blast_records.program
'blastn'
>>> blast_records.version
'BLASTN 2.2.27+'
>>> blast_records.reference
'Stephen F. Altschul, Thomas L. Madden, Alejandro A. Schäffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25:3389-3402.'
>>> blast_records.db
'refseq_rna'
>>> blast_records.param
{'expect': 10.0, 'sc-match': 2, 'sc-mismatch': -3, 'gap-open': 5, 'gap-extend': 2, 'filter': 'L;m;'}
>>> print(blast_records)
Program: BLASTN 2.2.27+
db: refseq_rna
Query: 42291 (length=61)
mystery_seq
Hits: ---- ----- ----------------------------------------------------------
# # HSP ID + description
---- ----- ----------------------------------------------------------
0 1 gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 52...
1 1 gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA...
2 1 gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA ...
3 2 gi|301171322|ref|NR_035857.1| Pan troglodytes microRNA...
...
The BLAST Record class
A Bio.Blast.Record object stores the information provided by BLAST for a
single query. The Bio.Blast.Record class inherits from list, and is
essentially a list of Bio.Blast.Hit objects (see section
The BLAST Hit class).
A Bio.Blast.Record object has the following two attributes:
query: ASeqRecordobject which may contain some or all of the following information:query.id: SeqId of the query;query.description: Definition line of the query;query.seq: The query sequence.
stat: A dictionary with statistical data of the BLAST hit. You may find the following keys in this dictionary:'db-num': number of sequences in BLAST db (integer);'db-len': length of BLAST db (integer);'hsp-len': effective HSP (High Scoring Pair) length (integer);'eff-space': effective search space (float);'kappa': Karlin-Altschul parameter K (float);'lambda': Karlin-Altschul parameter Lambda (float);'entropy': Karlin-Altschul parameter H (float)
message: Some (error?) information.
Continuing with our example,
>>> blast_record
<Bio.Blast.Record query.id='42291'; 100 hits>
>>> blast_record.query
SeqRecord(seq=Seq(None, length=61), id='42291', name='<unknown name>', description='mystery_seq', dbxrefs=[])
>>> blast_record.stat
{'db-num': 3056429, 'db-len': 673143725, 'hsp-len': 0, 'eff-space': 0, 'kappa': 0.41, 'lambda': 0.625, 'entropy': 0.78}
>>> print(blast_record)
Query: 42291 (length=61)
mystery_seq
Hits: ---- ----- ----------------------------------------------------------
# # HSP ID + description
---- ----- ----------------------------------------------------------
0 1 gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 52...
1 1 gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA...
2 1 gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA ...
3 2 gi|301171322|ref|NR_035857.1| Pan troglodytes microRNA...
...
As the Bio.Blast.Record class inherits from list, you can use it as
such. For example, you can iterate over the record:
>>> for hit in blast_record:
... hit
...
<Bio.Blast.Hit target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 1 HSP>
<Bio.Blast.Hit target.id='gi|301171311|ref|NR_035856.1|' query.id='42291'; 1 HSP>
<Bio.Blast.Hit target.id='gi|270133242|ref|NR_032573.1|' query.id='42291'; 1 HSP>
<Bio.Blast.Hit target.id='gi|301171322|ref|NR_035857.1|' query.id='42291'; 2 HSPs>
<Bio.Blast.Hit target.id='gi|301171267|ref|NR_035851.1|' query.id='42291'; 1 HSP>
...
To check how many hits the blast_record has, you can simply invoke Python’s
len function:
>>> len(blast_record)
100
Like Python lists, you can retrieve hits from a Bio.Blast.Record using
indices:
>>> blast_record[0] # retrieves the top hit
<Bio.Blast.Hit target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 1 HSP>
>>> blast_record[-1] # retrieves the last hit
<Bio.Blast.Hit target.id='gi|397513516|ref|XM_003827011.1|' query.id='42291'; 1 HSP>
To retrieve multiple hits from a Bio.Blast.Record, you can use the slice
notation. This will return a new Bio.Blast.Record object containing only
the sliced hits:
>>> blast_slice = blast_record[:3] # slices the first three hits
>>> print(blast_slice)
Query: 42291 (length=61)
mystery_seq
Hits: ---- ----- ----------------------------------------------------------
# # HSP ID + description
---- ----- ----------------------------------------------------------
0 1 gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 52...
1 1 gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA...
2 1 gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA ...
To create a copy of the Bio.Blast.Record, take the full slice:
>>> blast_record_copy = blast_record[:]
>>> type(blast_record_copy)
<class 'Bio.Blast.Record'>
>>> blast_record_copy # list of all hits
<Bio.Blast.Record query.id='42291'; 100 hits>
This is particularly useful if you want to sort or filter the BLAST record (see Sorting and filtering BLAST output), but want to retain a copy of the original BLAST output.
You can also access blast_record as a Python dictionary and retrieve hits
using the hit’s ID as key:
>>> blast_record["gi|262205317|ref|NR_030195.1|"]
<Bio.Blast.Hit target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 1 HSP>
If the ID is not found in the blast_record, a KeyError is raised:
>>> blast_record["unicorn_gene"]
Traceback (most recent call last):
...
KeyError: 'unicorn_gene'
You can get the full list of keys by using .keys() as usual:
>>> blast_record.keys()
['gi|262205317|ref|NR_030195.1|', 'gi|301171311|ref|NR_035856.1|', 'gi|270133242|ref|NR_032573.1|', ...]
What if you just want to check whether a particular hit is present in the query
results? You can do a simple Python membership test using the in keyword:
>>> "gi|262205317|ref|NR_030195.1|" in blast_record
True
>>> "gi|262205317|ref|NR_030194.1|" in blast_record
False
Sometimes, knowing whether a hit is present is not enough; you also want to
know the rank of the hit. Here, the index method comes to the rescue:
>>> blast_record.index("gi|301171437|ref|NR_035870.1|")
22
Remember that Python uses zero-based indexing, so the first hit will be at index 0.
The BLAST Hit class
Each Bio.Blast.Hit object in the blast_record list represents one BLAST
hit of the query against a target.
>>> hit = blast_record[0]
>>> hit
<Bio.Blast.Hit target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 1 HSP>
>>> hit.target
SeqRecord(seq=Seq(None, length=61), id='gi|262205317|ref|NR_030195.1|', name='NR_030195', description='Homo sapiens microRNA 520b (MIR520B), microRNA', dbxrefs=[])
We can get a summary of the hit by printing it:
>>> print(blast_record[3])
Query: 42291
mystery_seq
Hit: gi|301171322|ref|NR_035857.1| (length=86)
Pan troglodytes microRNA mir-520c (MIR520C), microRNA
HSPs: ---- -------- --------- ------ --------------- ---------------------
# E-value Bit score Span Query range Hit range
---- -------- --------- ------ --------------- ---------------------
0 8.9e-20 100.47 60 [1:61] [13:73]
1 3.3e-06 55.39 60 [0:60] [73:13]
You see that we’ve got the essentials covered here:
A hit is always for one query; the query ID and description are shown at the top of the summary.
A hit consists of one or more alignments of the query against one target sequence. The target information is shown next is the summary. As shown above, the target can be accessed via the
targetattribute of the hit.Finally, there’s a table containing quick information about the alignments each hit contains. In BLAST parlance, these alignments are called “High-scoring Segment Pairs”, or HSPs (see section The BLAST HSP class). Each row in the table summarizes one HSP, including the HSP index, e-value, bit score, span (the alignment length including gaps), query coordinates, and target coordinates.
The Bio.Blast.Hit class is a subclass of Bio.Align.Alignments (plural;
see Section The Alignments class), and therefore in essence is a list of
Bio.Align.Alignment (singular; see Section Alignment objects)
objects. In particular when aligning nucleotide sequences against the genome,
the Bio.Blast.Hit object may consist of more than one
Bio.Align.Alignment if a particular query aligns to more than one region of
a chromosome. For protein alignments, usually a hit consists of only one
alignment, especially for alignments of highly homologous sequences.
>>> type(hit)
<class 'Bio.Blast.Hit'>
>>> from Bio.Align import Alignments
>>> isinstance(hit, Alignments)
True
>>> len(hit)
1
For BLAST output in the XML2 format, a hit may have several targets with
identical sequences but different sequence IDs and descriptions. These targets
are accessible as the hit.targets attribute. In most cases, hit.targets
has length 1 and only contains hit.target:
>>> from Bio import Blast
>>> blast_record = Blast.read("xml_2900_blastx_001_v2.xml")
>>> for hit in blast_record:
... print(len(hit.targets))
...
1
1
2
1
1
1
1
1
1
1
However, as you can see in the output above, the third hit has multiple targets.
>>> hit = blast_record[2]
>>> hit.targets[0].seq
Seq(None, length=246)
>>> hit.targets[1].seq
Seq(None, length=246)
>>> hit.targets[0].id
'gi|684409690|ref|XP_009175831.1|'
>>> hit.targets[1].id
'gi|663044098|gb|KER20427.1|'
>>> hit.targets[0].name
'XP_009175831'
>>> hit.targets[1].name
'KER20427'
>>> hit.targets[0].description
'hypothetical protein T265_11027 [Opisthorchis viverrini]'
>>> hit.targets[1].description
'hypothetical protein T265_11027 [Opisthorchis viverrini]'
As the sequence contents for the two targets are identical to each other, their
sequence alignments are also identical. The alignments for this hit therefore
only refers to hit.targets[0] (which is identical to hit.target), as
the alignment for hit.targets[1] would be the same anyway.
The BLAST HSP class
Let’s return to our main example, and look at the first (and only) alignment in
the first hit. This alignment is an instance of the Bio.Blast.HSP class,
which is a subclass of the Alignment class in Bio.Align:
>>> from Bio import Blast
>>> blast_record = Blast.read("my_blast.xml")
>>> hit = blast_record[0]
>>> len(hit)
1
>>> alignment = hit[0]
>>> alignment
<Bio.Blast.HSP target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 2 rows x 61 columns>
>>> type(alignment)
<class 'Bio.Blast.HSP'>
>>> from Bio.Align import Alignment
>>> isinstance(alignment, Alignment)
True
The alignment object has attributes pointing to the target and query
sequences, as well as a coordinates attribute describing the sequence
alignment.
>>> alignment.target
SeqRecord(seq=Seq('CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTT...GGG'), id='gi|262205317|ref|NR_030195.1|', name='NR_030195', description='Homo sapiens microRNA 520b (MIR520B), microRNA', dbxrefs=[])
>>> alignment.query
SeqRecord(seq=Seq('CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTT...GGG'), id='42291', name='<unknown name>', description='mystery_seq', dbxrefs=[])
>>> alignment.target
SeqRecord(seq=Seq('CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTT...GGG'), id='gi|262205317|ref|NR_030195.1|', name='NR_030195', description='Homo sapiens microRNA 520b (MIR520B), microRNA', dbxrefs=[])
>>> alignment.query
SeqRecord(seq=Seq('CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTT...GGG'), id='42291', name='<unknown name>', description='mystery_seq', dbxrefs=[])
>>> print(alignment.coordinates)
[[ 0 61]
[ 0 61]]
For translated BLAST searches, the features attribute of the target or
query may contain a SeqFeature of type CDS that stores the amino acid
sequence region. The qualifiers attribute of such a feature is a
dictionary with a single key 'coded_by'; the corresponding value specifies
the nucleotide sequence region, in a GenBank-style string with 1-based
coordinates, that encodes the amino acid sequence.
Each Alignment object has the following additional attributes:
score: score of the High Scoring Pair (HSP);annotations: a dictionary that may contain the following keys:'bit score': score (in bits) of HSP (float);'evalue': e-value of HSP (float);'identity’: number of identities in HSP (integer);'positive': number of positives in HSP (integer);'gaps': number of gaps in HSP (integer);'midline': formatting middle line.
The usual Alignment methods (see Section Alignment objects) can be
applied to the alignment. For example, we can print the alignment:
>>> print(alignment)
Query : 42291 Length: 61 Strand: Plus
mystery_seq
Target: gi|262205317|ref|NR_030195.1| Length: 61 Strand: Plus
Homo sapiens microRNA 520b (MIR520B), microRNA
Score:111 bits(122), Expect:5e-23,
Identities:61/61(100%), Positives:61/61(100%), Gaps:0.61(0%)
gi|262205 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTTTAGAGG
0 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
42291 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTTTAGAGG
gi|262205 60 G 61
60 | 61
42291 60 G 61
Let’s just print out some summary info about all hits in our BLAST record greater than a particular threshold:
>>> E_VALUE_THRESH = 0.04
>>> for alignments in blast_record:
... for alignment in alignments:
... if alignment.annotations["evalue"] < E_VALUE_THRESH:
... print("****Alignment****")
... print("sequence:", alignment.target.id, alignment.target.description)
... print("length:", len(alignment.target))
... print("score:", alignment.score)
... print("e value:", alignment.annotations["evalue"])
... print(alignment[:, :50])
...
****Alignment****
sequence: gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 520b (MIR520B), microRNA
length: 61
score: 122.0
e value: 4.91307e-23
gi|262205 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTC 50
0 |||||||||||||||||||||||||||||||||||||||||||||||||| 50
42291 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTC 50
****Alignment****
sequence: gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA mir-520b (MIR520B), microRNA
length: 60
score: 120.0
e value: 1.71483e-22
gi|301171 0 CCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCC 50
0 |||||||||||||||||||||||||||||||||||||||||||||||||| 50
42291 1 CCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCC 51
****Alignment****
sequence: gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA mir-519a (MIR519A), microRNA
length: 85
score: 112.0
e value: 2.54503e-20
gi|270133 12 CCCTCTAGAGGGAAGCGCTTTCTGTGGTCTGAAAGAAAAGAAAGTGCTTC 62
0 |||||||.|||||||||||||||||.|||||||||||||||||||||||| 50
42291 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTC 50
...
Sorting and filtering BLAST output
If the ordering of hits in the BLAST output file doesn’t suit your taste, you
can use the sort method to resort the hits in the Bio.Blast.Record
object. As an example, here we sort the hits based on the sequence length of
each target, setting the reverse flag to True so that we sort in
descending order.
>>> for hit in blast_record[:5]:
... print(f"{hit.target.id} {len(hit.target)}")
...
gi|262205317|ref|NR_030195.1| 61
gi|301171311|ref|NR_035856.1| 60
gi|270133242|ref|NR_032573.1| 85
gi|301171322|ref|NR_035857.1| 86
gi|301171267|ref|NR_035851.1| 80
>>> sort_key = lambda hit: len(hit.target)
>>> blast_record.sort(key=sort_key, reverse=True)
>>> for hit in blast_recordt[:5]:
... print(f"{hit.target.id} {len(hit.target)}")
...
gi|397513516|ref|XM_003827011.1| 6002
gi|390332045|ref|XM_776818.2| 4082
gi|390332043|ref|XM_003723358.1| 4079
gi|356517317|ref|XM_003527287.1| 3251
gi|356543101|ref|XM_003539954.1| 2936
This will sort blast_record in place.
Use original_blast_record = blast_record[:] before sorting if you want to
retain a copy of the original, unsorted BLAST output.
To filter BLAST hits based on their properties, you can use Python’s built-in
filter with the approciate callback function to evaluate each hit. The
callback function must accept as its argument a single Hit object and
return True or False. Here is an example in which we filter out
Hit objects that only have one HSP:
>>> filter_func = lambda hit: len(hit) > 1 # the callback function
>>> len(blast_record) # no. of hits before filtering
100
>>> blast_record[:] = filter(filter_func, blast_record)
>>> len(blast_record) # no. of hits after filtering
37
>>> for hit in blast_record[:5]: # quick check for the hit lengths
... print(f"{hit.target.id} {len(hit)}")
...
gi|301171322|ref|NR_035857.1| 2
gi|262205330|ref|NR_030198.1| 2
gi|301171447|ref|NR_035871.1| 2
gi|262205298|ref|NR_030190.1| 2
gi|270132717|ref|NR_032716.1| 2
Similarly, you can filter HSPs in each hit, for example on their e-value:
>>> filter_func = lambda hsp: hsp.annotations["evalue"] < 1.0e-12
>>> for hit in blast_record:
... hit[:] = filter(filter_func, hit)
...
Probably you’d want to follow this up by removing all hits with no HSPs remaining:
>>> filter_func = lambda hit: len(hit) > 0
>>> blast_record[:] = filter(filter_func, blast_record)
>>> len(blast_record)
16
Use Python’s built-in map function to modify hits or HSPs in the BLAST
record. The map function accepts a callback function returning the modified
hit object. For example, we can use map to rename the hit IDs:
>>> for hit in blast_record[:5]:
... print(hit.target.id)
...
gi|301171322|ref|NR_035857.1|
gi|262205330|ref|NR_030198.1|
gi|301171447|ref|NR_035871.1|
gi|262205298|ref|NR_030190.1|
gi|270132717|ref|NR_032716.1|
>>> import copy
>>> original_blast_record = copy.deepcopy(blast_record)
>>> def map_func(hit):
... # renames "gi|301171322|ref|NR_035857.1|" to "NR_035857.1"
... hit.target.id = hit.target.id.split("|")[3]
... return hit
...
>>> blast_record[:] = map(map_func, blast_record)
>>> for hit in blast_record[:5]:
... print(hit.target.id)
...
NR_035857.1
NR_030198.1
NR_035871.1
NR_030190.1
NR_032716.1
>>> for hit in original_blast_record[:5]:
... print(hit.target.id)
...
gi|301171322|ref|NR_035857.1|
gi|262205330|ref|NR_030198.1|
gi|301171447|ref|NR_035871.1|
gi|262205298|ref|NR_030190.1|
gi|270132717|ref|NR_032716.1|
Note that in this example, map_func modifies the hit in-place.
In contrast to sorting and filtering (see above), using
original_blast_record = blast_record[:] is not sufficient to retain a copy
of the unmodified BLAST record, as it creates a shallow copy of the BLAST
record, consisting of pointers to the same Hit objects. Instead, we use
copy.deepcopy to create a copy of the BLAST record in which each Hit
object is duplicated.
Writing BLAST records
Use the write function in Bio.Blast to save BLAST records as an XML
file. By default, the (DTD-based) XML format is used; you can also save the
BLAST records in the (schema-based) XML2 format by using the fmt="XML2"
argument to the write function.
>>> from Bio import Blast
>>> stream = Blast.qblast("blastn", "nt", "8332116")
>>> records = Blast.parse(stream)
>>> Blast.write(records, "my_qblast_output.xml")
or
>>> Blast.write(records, "my_qblast_output.xml", fmt="XML2")
In this example, we could have saved the data returned by Blast.qblast
directly to an XML file (see section Saving BLAST results).
However, by parsing the data returned by qblast into records, we can sort or
filter the BLAST records before saving them. For example, we may be interested
only in BLAST HSPs with a positive score of at least 400:
>>> filter_func = lambda hsp: hsp.annotations["positive"] >= 400
>>> for hit in records[0]:
... hit[:] = filter(filter_func, hit)
...
>>> Blast.write(records, "my_qblast_output_selected.xml")
Instead of a file name, the second argument to Blast.write can also be a
file stream. In that case, the stream must be opened in binary format for
writing:
>>> with open("my_qblast_output.xml", "wb") as stream:
... Blast.write(records, stream)
...
Dealing with PSI-BLAST
You can run the standalone version of PSI-BLAST (psiblast) directly
from the command line or using python’s subprocess module.
At the time of writing, the NCBI do not appear to support tools running a PSI-BLAST search via the internet.
Note that the Bio.Blast parser can read the XML output from current
versions of PSI-BLAST, but information like which sequences in each
iteration is new or reused isn’t present in the XML file.
Dealing with RPS-BLAST
You can run the standalone version of RPS-BLAST (rpsblast) directly
from the command line or using python’s subprocess module.
At the time of writing, the NCBI do not appear to support tools running an RPS-BLAST search via the internet.
You can use the Bio.Blast parser to read the XML output from current
versions of RPS-BLAST.
BLAST (old)
Hey, everybody loves BLAST right? I mean, geez, how can it get any easier to do comparisons between one of your sequences and every other sequence in the known world? But, of course, this section isn’t about how cool BLAST is, since we already know that. It is about the problem with BLAST – it can be really difficult to deal with the volume of data generated by large runs, and to automate BLAST runs in general.
Fortunately, the Biopython folks know this only too well, so they’ve developed lots of tools for dealing with BLAST and making things much easier. This section details how to use these tools and do useful things with them.
Dealing with BLAST can be split up into two steps, both of which can be done from within Biopython. Firstly, running BLAST for your query sequence(s), and getting some output. Secondly, parsing the BLAST output in Python for further analysis.
Your first introduction to running BLAST was probably via the NCBI web-service. In fact, there are lots of ways you can run BLAST, which can be categorized in several ways. The most important distinction is running BLAST locally (on your own machine), and running BLAST remotely (on another machine, typically the NCBI servers). We’re going to start this chapter by invoking the NCBI online BLAST service from within a Python script.
NOTE: The following Chapter BLAST and other sequence search tools
describes Bio.SearchIO. We intend this to ultimately replace the
older Bio.Blast module, as it provides a more general framework
handling other related sequence searching tools as well. However, for
now you can use either that or the older Bio.Blast module for
dealing with NCBI BLAST.
Running BLAST over the Internet
We use the function qblast() in the Bio.Blast.NCBIWWW module to
call the online version of BLAST. This has three non-optional arguments:
The first argument is the blast program to use for the search, as a lower case string. The options and descriptions of the programs are available at https://blast.ncbi.nlm.nih.gov/Blast.cgi. Currently
qblastonly works with blastn, blastp, blastx, tblast and tblastx.The second argument specifies the databases to search against. Again, the options for this are available on the NCBI Guide to BLAST https://blast.ncbi.nlm.nih.gov/doc/blast-help/.
The third argument is a string containing your query sequence. This can either be the sequence itself, the sequence in fasta format, or an identifier like a GI number.
The NCBI guidelines, from https://blast.ncbi.nlm.nih.gov/doc/blast-help/developerinfo.html#developerinfo state:
Do not contact the server more often than once every 10 seconds.
Do not poll for any single RID more often than once a minute.
Use the URL parameter email and tool, so that the NCBI can contact you if there is a problem.
Run scripts weekends or between 9 pm and 5 am Eastern time on weekdays if more than 50 searches will be submitted.
To fulfill the third point, one can set the NCBIWWW.email variable.
>>> from Bio.Blast import NCBIWWW
>>> NCBIWWW.email = "A.N.Other@example.com"
The qblast function also takes a number of other option arguments,
which are basically analogous to the different parameters you can set on
the BLAST web page. We’ll just highlight a few of them here:
The argument
url_basesets the base URL for running BLAST over the internet. By default it connects to the NCBI.The
qblastfunction can return the BLAST results in various formats, which you can choose with the optionalformat_typekeyword:"HTML","Text","ASN.1", or"XML". The default is"XML", as that is the format expected by the parser, described in section Parsing BLAST output below.The argument
expectsets the expectation or e-value threshold.
For more about the optional BLAST arguments, we refer you to the NCBI’s own documentation, or that built into Biopython:
>>> from Bio.Blast import NCBIWWW
>>> help(NCBIWWW.qblast)
Note that the default settings on the NCBI BLAST website are not quite the same as the defaults on QBLAST. If you get different results, you’ll need to check the parameters (e.g., the expectation value threshold and the gap values).
For example, if you have a nucleotide sequence you want to search against the nucleotide database (nt) using BLASTN, and you know the GI number of your query sequence, you can use:
>>> from Bio.Blast import NCBIWWW
>>> result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
Alternatively, if we have our query sequence already in a FASTA formatted file, we just need to open the file and read in this record as a string, and use that as the query argument:
>>> from Bio.Blast import NCBIWWW
>>> fasta_string = open("m_cold.fasta").read()
>>> result_handle = NCBIWWW.qblast("blastn", "nt", fasta_string)
We could also have read in the FASTA file as a SeqRecord and then
supplied just the sequence itself:
>>> from Bio.Blast import NCBIWWW
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", format="fasta")
>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.seq)
Supplying just the sequence means that BLAST will assign an identifier
for your sequence automatically. You might prefer to use the
SeqRecord object’s format method to make a FASTA string (which will
include the existing identifier):
>>> from Bio.Blast import NCBIWWW
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", format="fasta")
>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.format("fasta"))
This approach makes more sense if you have your sequence(s) in a
non-FASTA file format which you can extract using Bio.SeqIO (see
Chapter Sequence Input/Output).
Whatever arguments you give the qblast() function, you should get
back your results in a handle object (by default in XML format). The
next step would be to parse the XML output into Python objects
representing the search results (Section Parsing BLAST output),
but you might want to save a local copy of the output file first. I find
this especially useful when debugging my code that extracts info from
the BLAST results (because re-running the online search is slow and
wastes the NCBI computer time).
We need to be a bit careful since we can use result_handle.read() to
read the BLAST output only once – calling result_handle.read() again
returns an empty string.
>>> with open("my_blast.xml", "w") as out_handle:
... out_handle.write(result_handle.read())
...
>>> result_handle.close()
After doing this, the results are in the file my_blast.xml and the
original handle has had all its data extracted (so we closed it).
However, the parse function of the BLAST parser (described
in Parsing BLAST output) takes a file-handle-like object, so we
can just open the saved file for input:
>>> result_handle = open("my_blast.xml")
Now that we’ve got the BLAST results back into a handle again, we are ready to do something with them, so this leads us right into the parsing section (see Section Parsing BLAST output below). You may want to jump ahead to that now ….
Running BLAST locally
Introduction
Running BLAST locally (as opposed to over the internet, see Section Running BLAST over the Internet) has at least major two advantages:
Local BLAST may be faster than BLAST over the internet;
Local BLAST allows you to make your own database to search for sequences against.
Dealing with proprietary or unpublished sequence data can be another reason to run BLAST locally. You may not be allowed to redistribute the sequences, so submitting them to the NCBI as a BLAST query would not be an option.
Unfortunately, there are some major drawbacks too – installing all the bits and getting it setup right takes some effort:
Local BLAST requires command line tools to be installed.
Local BLAST requires (large) BLAST databases to be setup (and potentially kept up to date).
To further confuse matters there are several different BLAST packages available, and there are also other tools which can produce imitation BLAST output files, such as BLAT.
Standalone NCBI BLAST+
The “new” NCBI BLAST+ suite was released in 2009. This replaces the old NCBI “legacy” BLAST package (see below).
This section will show briefly how to use these tools from within Python. If you have already read or tried the alignment tool examples in Section Alignment Tools this should all seem quite straightforward. First, we construct a command line string (as you would type in at the command line prompt if running standalone BLAST by hand). Then we can execute this command from within Python.
For example, taking a FASTA file of gene nucleotide sequences, you might want to run a BLASTX (translation) search against the non-redundant (NR) protein database. Assuming you (or your systems administrator) has downloaded and installed the NR database, you might run:
$ blastx -query opuntia.fasta -db nr -out opuntia.xml -evalue 0.001 -outfmt 5
This should run BLASTX against the NR database, using an expectation cut-off value of \(0.001\) and produce XML output to the specified file (which we can then parse). On my computer this takes about six minutes - a good reason to save the output to a file so you can repeat any analysis as needed.
From within python we can use the subprocess module to build the
command line string, and run it:
>>> import subprocess
>>> cmd = "blastx -query opuntia.fasta -db nr -out opuntia.xml"
>>> cmd += " -evalue 0.001 -outfmt 5"
>>> subprocess.run(cmd, shell=True)
In this example there shouldn’t be any output from BLASTX to the
terminal. You may want to check the output file opuntia.xml has been
created.
As you may recall from earlier examples in the tutorial, the
opuntia.fasta contains seven sequences, so the BLAST XML output
should contain multiple results. Therefore use
Bio.Blast.NCBIXML.parse() to parse it as described below in
Section Parsing BLAST output.
Other versions of BLAST
NCBI BLAST+ (written in C++) was first released in 2009 as a replacement
for the original NCBI “legacy” BLAST (written in C) which is no longer
being updated. There were a lot of changes – the old version had a
single core command line tool blastall which covered multiple
different BLAST search types (which are now separate commands in
BLAST+), and all the command line options were renamed. Biopython’s
wrappers for the NCBI “legacy” BLAST tools have been deprecated and will
be removed in a future release. To try to avoid confusion, we do not
cover calling these old tools from Biopython in this tutorial.
You may also come across Washington University
BLAST (WU-BLAST), and its successor,
Advanced Biocomputing BLAST
(AB-BLAST, released in 2009, not free/open source). These packages
include the command line tools wu-blastall and ab-blastall,
which mimicked blastall from the NCBI “legacy” BLAST suite.
Biopython does not currently provide wrappers for calling these tools,
but should be able to parse any NCBI compatible output from them.
Parsing BLAST output
As mentioned above, BLAST can generate output in various formats, such
as XML, HTML, and plain text. Originally, Biopython had parsers for
BLAST plain text and HTML output, as these were the only output formats
offered at the time. Unfortunately, the BLAST output in these formats
kept changing, each time breaking the Biopython parsers. Our HTML BLAST
parser has been removed, while the deprecated plain text BLAST parser is
now only available via Bio.SearchIO. Use it at your own risk, it may
or may not work, depending on which BLAST version you’re using.
As keeping up with changes in BLAST became a hopeless endeavor, especially with users running different BLAST versions, we now recommend to parse the output in XML format, which can be generated by recent versions of BLAST. Not only is the XML output more stable than the plain text and HTML output, it is also much easier to parse automatically, making Biopython a whole lot more stable.
You can get BLAST output in XML format in various ways. For the parser, it doesn’t matter how the output was generated, as long as it is in the XML format.
You can use Biopython to run BLAST over the internet, as described in section Running BLAST over the Internet.
You can use Biopython to run BLAST locally, as described in section Running BLAST locally.
You can do the BLAST search yourself on the NCBI site through your web browser, and then save the results. You need to choose XML as the format in which to receive the results, and save the final BLAST page you get (you know, the one with all of the interesting results!) to a file.
You can also run BLAST locally without using Biopython, and save the output in a file. Again, you need to choose XML as the format in which to receive the results.
The important point is that you do not have to use Biopython scripts to
fetch the data in order to be able to parse it. Doing things in one of
these ways, you then need to get a handle to the results. In Python, a
handle is just a nice general way of describing input to any info source
so that the info can be retrieved using read() and readline()
functions (see
Section What the heck is a handle?).
If you followed the code above for interacting with BLAST through a
script, then you already have result_handle, the handle to the BLAST
results. For example, using a GI number to do an online search:
>>> from Bio.Blast import NCBIWWW
>>> result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
If instead you ran BLAST some other way, and have the BLAST output (in
XML format) in the file my_blast.xml, all you need to do is to open
the file for reading:
>>> result_handle = open("my_blast.xml")
Now that we’ve got a handle, we are ready to parse the output. The code to parse it is really quite small. If you expect a single BLAST result (i.e., you used a single query):
>>> from Bio.Blast import NCBIXML
>>> blast_record = NCBIXML.read(result_handle)
or, if you have lots of results (i.e., multiple query sequences):
>>> from Bio.Blast import NCBIXML
>>> blast_records = NCBIXML.parse(result_handle)
Just like Bio.SeqIO and Bio.Align (see
Chapters Sequence Input/Output
and Sequence alignments), we have a pair of input
functions, read and parse, where read is for when you have
exactly one object, and parse is an iterator for when you can have
lots of objects – but instead of getting SeqRecord or
MultipleSeqAlignment objects, we get BLAST record objects.
To be able to handle the situation where the BLAST file may be huge,
containing thousands of results, NCBIXML.parse() returns an
iterator. In plain English, an iterator allows you to step through the
BLAST output, retrieving BLAST records one by one for each BLAST search
result:
>>> from Bio.Blast import NCBIXML
>>> blast_records = NCBIXML.parse(result_handle)
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
StopIteration
# No further records
Or, you can use a for-loop:
>>> for blast_record in blast_records:
... pass # Do something with blast_record
...
Note though that you can step through the BLAST records only once. Usually, from each BLAST record you would save the information that you are interested in. If you want to save all returned BLAST records, you can convert the iterator into a list:
>>> blast_records = list(blast_records)
Now you can access each BLAST record in the list with an index as usual. If your BLAST file is huge though, you may run into memory problems trying to save them all in a list.
Usually, you’ll be running one BLAST search at a time. Then, all you
need to do is to pick up the first (and only) BLAST record in
blast_records:
>>> from Bio.Blast import NCBIXML
>>> blast_records = NCBIXML.parse(result_handle)
>>> blast_record = next(blast_records)
or more elegantly:
>>> from Bio.Blast import NCBIXML
>>> blast_record = NCBIXML.read(result_handle)
I guess by now you’re wondering what is in a BLAST record.
The BLAST record class
A BLAST Record contains everything you might ever want to extract from the BLAST output. Right now we’ll just show an example of how to get some info out of the BLAST report, but if you want something in particular that is not described here, look at the info on the record class in detail, and take a gander into the code or automatically generated documentation – the docstrings have lots of good info about what is stored in each piece of information.
To continue with our example, let’s just print out some summary info about all hits in our blast report greater than a particular threshold. The following code does this:
>>> E_VALUE_THRESH = 0.04
>>> for alignment in blast_record.alignments:
... for hsp in alignment.hsps:
... if hsp.expect < E_VALUE_THRESH:
... print("****Alignment****")
... print("sequence:", alignment.title)
... print("length:", alignment.length)
... print("e value:", hsp.expect)
... print(hsp.query[0:75] + "...")
... print(hsp.match[0:75] + "...")
... print(hsp.sbjct[0:75] + "...")
...
This will print out summary reports like the following:
****Alignment****
sequence: >gb|AF283004.1|AF283004 Arabidopsis thaliana cold acclimation protein WCOR413-like protein
alpha form mRNA, complete cds
length: 783
e value: 0.034
tacttgttgatattggatcgaacaaactggagaaccaacatgctcacgtcacttttagtcccttacatattcctc...
||||||||| | ||||||||||| || |||| || || |||||||| |||||| | | |||||||| ||| ||...
tacttgttggtgttggatcgaaccaattggaagacgaatatgctcacatcacttctcattccttacatcttcttc...
Basically, you can do anything you want to with the info in the BLAST report once you have parsed it. This will, of course, depend on what you want to use it for, but hopefully this helps you get started on doing what you need to do!
An important consideration for extracting information from a BLAST
report is the type of objects that the information is stored in. In
Biopython, the parsers return Record objects, either Blast or
PSIBlast depending on what you are parsing. These objects are
defined in Bio.Blast.Record and are quite complete.
Figures Class diagram for the Blast Record class representing a BLAST report. and Class diagram for the PSIBlast Record class. and
are my attempts at UML class diagrams for the Blast and PSIBlast
record classes. The PSIBlast record object is similar, but has support
for the rounds that are used in the iteration steps of PSIBlast.
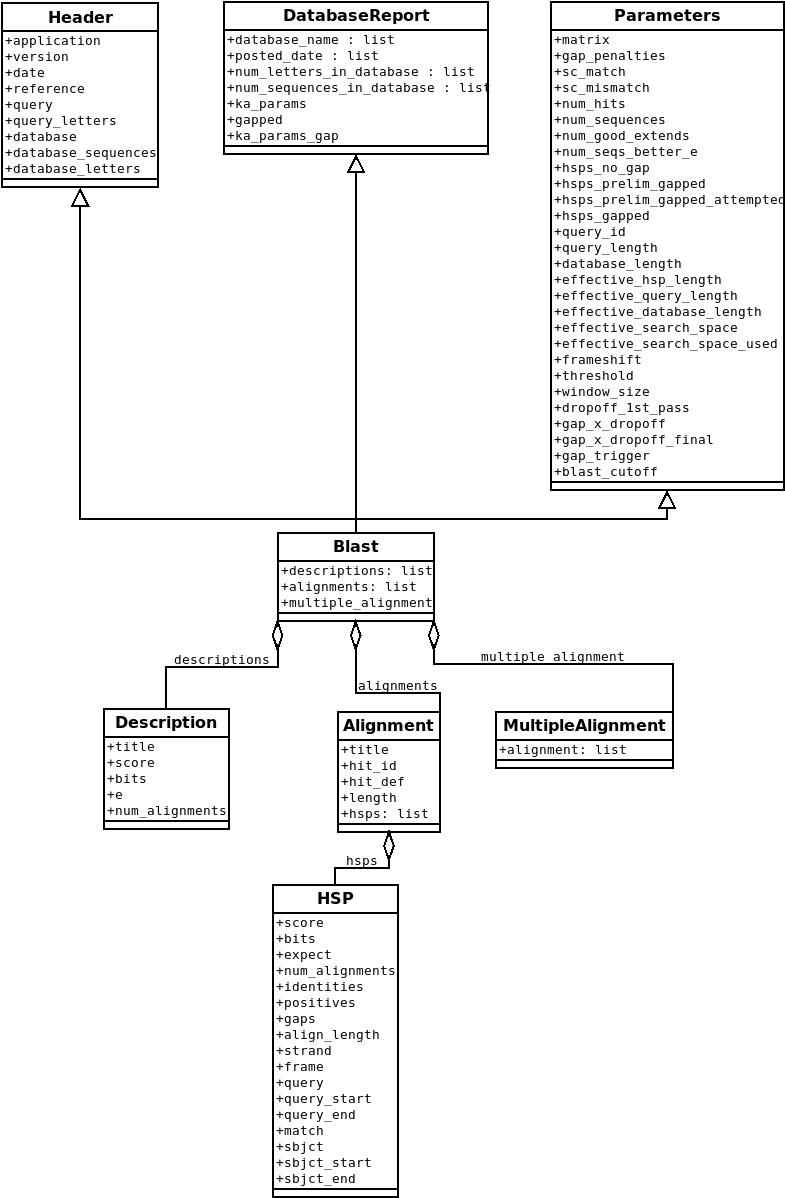
Fig. 1 Class diagram for the Blast Record class representing a BLAST report.
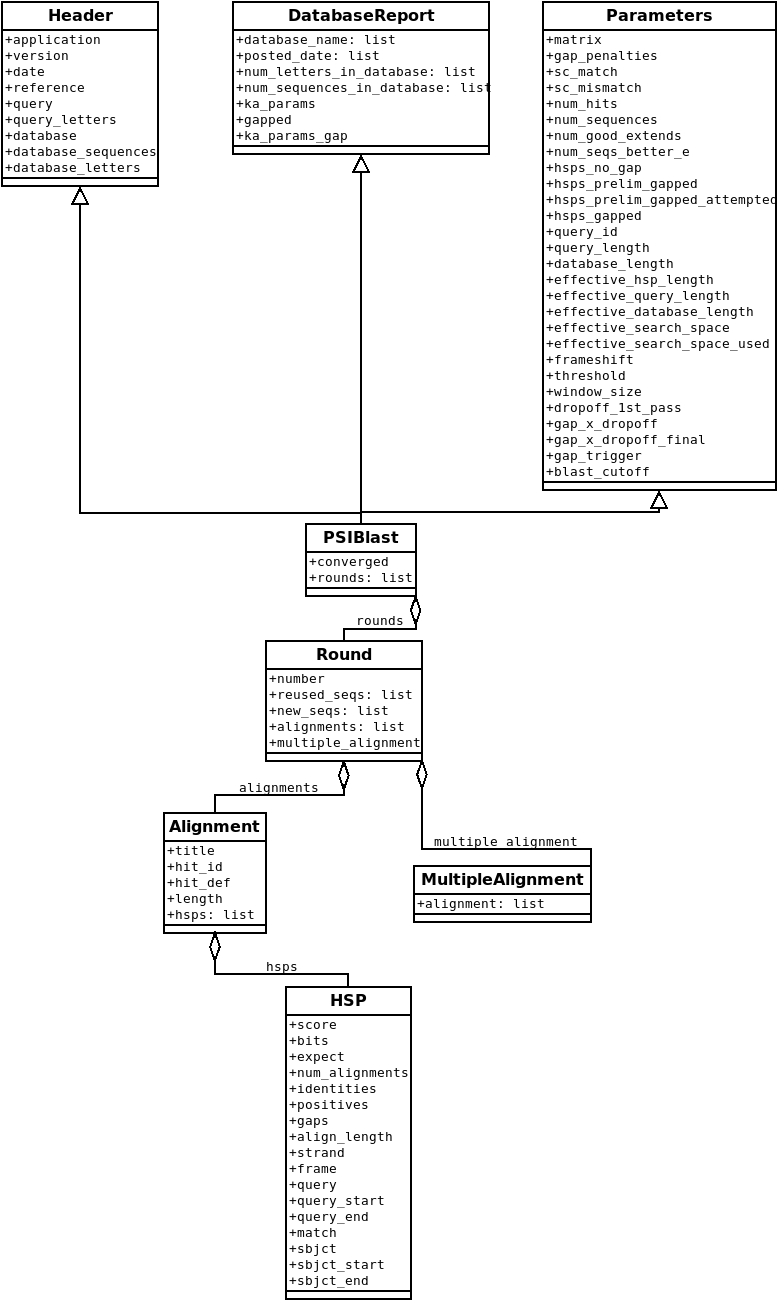
Fig. 2 Class diagram for the PSIBlast Record class.
If you are good at UML and see mistakes/improvements that can be made, please let me know.
Dealing with PSI-BLAST
You can run the standalone version of PSI-BLAST (the legacy NCBI command
line tool blastpgp, or its replacement psiblast) directly from
the command line or using python’s subprocess module.
At the time of writing, the NCBI do not appear to support tools running a PSI-BLAST search via the internet.
Note that the Bio.Blast.NCBIXML parser can read the XML output from
current versions of PSI-BLAST, but information like which sequences in
each iteration is new or reused isn’t present in the XML file.
Dealing with RPS-BLAST
You can run the standalone version of RPS-BLAST (either the legacy NCBI
command line tool rpsblast, or its replacement with the same name)
directly from the command line or using python’s subprocess module.
At the time of writing, the NCBI do not appear to support tools running an RPS-BLAST search via the internet.
You can use the Bio.Blast.NCBIXML parser to read the XML output from
current versions of RPS-BLAST.